Craniosynotosis Surgery
The skull is made up of several bone plates, which are separated from each other by small gaps acting as growth lines. These growth lines are called the cranial sutures and they coordinate growth between the brain and the skull so as the brain grows the skull grows with it. The sutures normally remain open and functional until adulthood. They are located in specific places in the skull and separate the frontal bones from the parietal bones and the occipital bone. Each suture has its own name - there are 2 coronal sutures; 2 lambdoid sutures; 1 metopic suture and 1 sagittal suture.
Craniosynostosis is the name given to a rare congenital abnormality affecting approximately 1 in 3,000 children causing premature or early fusion of one or more of the baby’s cranial sutures during pregnancy and causing the skull, and therefore head shape to grow abnormally. In addition to the appearance related issues, untreated cases may occasionally lead to pressure on the growing brain due to lack of space. This can potentially lead to significant regular headaches and in extreme cases, fits and seizures together with damage to vision and development. Different predictable head shapes are produced depending on which particular suture is fused.
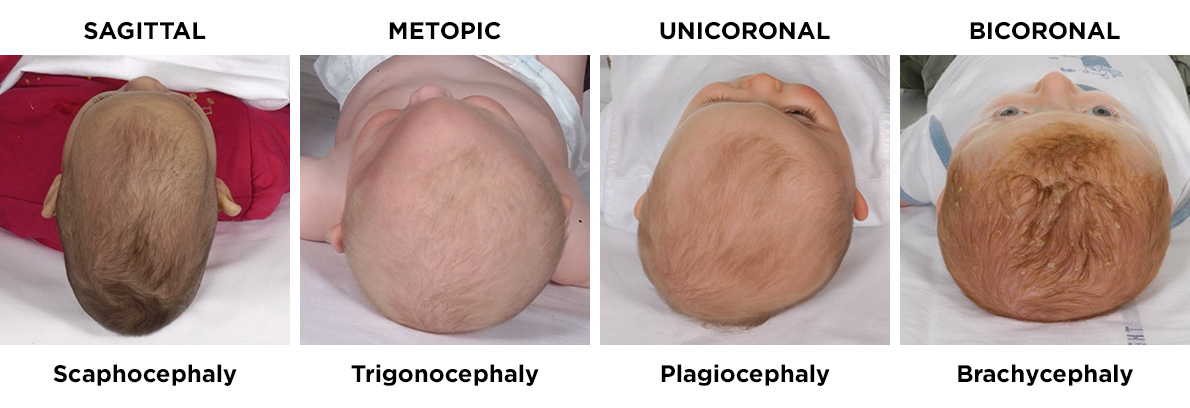
The above photographs show the typical head shapes produced by fusion of different major sutures. The name of the suture fused is on top of the photograph whilst the medical term for the resultant shape of the head is placed below the photograph.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
It is important that rare craniofacial conditions are treated in specialist centres which see large numbers of cases each year in order to achieve the best possible results. Mr Johnson is the leader and Director of the Oxford Craniofacial Unit, one of only 4 nationally designated highly specialised centres in the UK. Consequently children who have very complex conditions, many of whom would have not survived if they were born several years ago, can now be given the opportunity to fulfil their natural potential.

The management of craniosynostosis
The treatment of children with craniosynostosis is primarily surgical but the management of the affected children is multidisciplinary and involves a large team comprising many different specialists who follow these children up in the outpatient clinic until they are fully grown. The specialists involved in managing these children include Plastic surgeons, Maxillofacial surgeons, Neurosurgeons, ENT surgeons, Ophthalmic surgeons, Paediatric anaesthetists, Geneticists, Paediatricians, Orthodontists, Speech and Language therapists, Clinical psychologists, Orthoptists. Most of the cases are not genetic and can be corrected with a single operation but approximately 30% have a genetic cause and form part of a syndrome which can lead to very complex abnormalities of the whole face as well as other parts of the body including the hands and feet. There are well over 50 different syndromes associated with craniosynostosis. Children with syndromic forms of craniosynostosis often require multiple surgical procedures at different stages from birth to maturity.


Scroll to the bottom of the page to watch Mr Johnson and the team in the 3 part BBC 2 documentary on Childrens Craniofacial Surgery.
The surgical treatment of craniosynostosis
Craniofacial surgery to change the shape of the skull and increase space for the brain, in Oxford, is undertaken by a small number of dedicated and highly trained plastic surgeons and paediatric neurosurgeons together with specialist paediatric anaesthetists. This surgery is typically undertaken when children are between 12 – 18 months of age though occasionally surgery is needed much earlier and sometimes even within a few weeks of life. The operations essentially involve making a zig zag shaped scar across the top of the head within the hair from one ear to the other ear. Large pieces of bone are carefully removed and reshaped (by bending and cutting) and rearranged by essentially turning the top of the skull into a 3-dimensional jigsaw puzzle which is then replaced back into the head to improve the shape and create more space for the brain to grow. In some cases the surgery can be limited to reshaping the forehead (called a Frontoorbital Advancement and Remodelling procedure, or FOAR for short). In other cases the whole of the top portion of the skull can be shaped (called a Total Calvarial Remodelling procedure, or TCR for short). In cases of bicoronal synostosis surgery is performed to create more space for the brain by incrementally lengthening the back of the skull (called a Posterior Vault Distraction or PVD for short).
Surgery to advance the mid portion of the face (called a Le Fort III advancement) involves both plastic surgery and maxillofacial surgery. This procedure is undertaken to correct breathing difficulties and improve the appearance of the eyes and midface in patients with complex syndromes. Surgery to advance the midface can be combined with surgery to the forehead (called a Monobloc procedure). In Oxford this surgery usually requires children to have a rigid external distraction frame (called a RED frame) fitted at the time of the surgery and to keep the frame for a period of 2-3 months before it is removed.

Non Syndrominc Single Suture Craniosynostosis
Click on the links below to find out more about each of these procedures:
This is the commonest form of single suture synostoses and occurs in approximately 1 in 5,000 live births. The vast majority of cases are not genetic in origin and are most commonly caused by reduced space for the baby’s head in the uterus during pregnancy. A small number of cases may have a genetic cause. It is 4 times commoner in boys than girls, though no one really understands why. Sagittal synostosis leads to a head shape which is long and narrow (called scaphocephaly) with a tendency for the forehead to be prominent and bossed and the back of the head to be prominent and pointed. Surgery, is in the form of a Total Calvarial Remodelling procedure (TCR) which usually takes place when affected children are between 12-18 months of age (though the window of opportunity to undertake the surgery is quite wide). The aim of surgery is to stop the head shape from getting worse, make the head shape better by reshaping the whole of the top portion of the skull and finally to reduce the risk of a child with sagittal synostosis developing raised intracranial pressure from approximately 15% (in cases that do not have surgery) to less than 2% (in cases that have do have surgery) by creating more space around the brain.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
This is the second commonest suture to fuse and occurs in approximately 1 in 10,000 live births. The vast majority of cases are not genetic in origin and are most commonly caused by reduced space for the baby’s head in the uterus during pregnancy. A small number of cases have a genetic cause. It is commoner in boys than girls, though no one really understands why. Metopic synostosis leads to a forehead which is triangular in shape (called trigonocephaly) with a tendency for the eyes to appear closer together than normal. Surgery, is in the form of a Frontoorbital Advancement and Remodelling procedure (FOAR) which usually takes place when affected children are between 12-18 months of age (though the widow of opportunity to undertake the surgery is quite wide). The aim of surgery is to stop the head shape from getting worse, make the head shape better by reshaping the forehead and finally to reduce the risk of a child with metopic synostosis developing raised intracranial pressure from approximately 15% (in cases that do not have surgery) to less than 2% (in cases that have do have surgery) by creating more space around the brain.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Unicoronal synostosis refers to fusion of one of the 2 coronal sutures. It is more likely to be genetic in origin than the other single suture synostoses though some cases may still be caused by reduced space for the baby’s head in the uterus during pregnancy. Syndromes potentially causing unicoronal synostosis include Saethre Chotzen syndrome, Muenke syndrome and TCF 12 related craniosynostosis.
Unicoronal synostosis leads to a forehead which is flatter on the affected side (called plagiocephaly) with the affected eyebrow looking more elevated and the eye looking more widely open than the opposite side. The shape of the skull from above looks like a trapezium and there is a shortened distance from the ear to the eye on the affected side compared with the unaffected side. There is some associated facial asymmetry with the nose and the chin being deviated to the opposite side.
Surgery for unicoronal synostosis is in the form of a Frontoorbital Advancement and Remodelling procedure (FOAR) which usually takes place when affected children are between 12-18 months of age (though the widow of opportunity to undertake the surgery is quite wide). The aim of surgery is to stop the head shape from getting worse, make the head shape better by reshaping the forehead and finally to reduce the risk of a child with unicoronal synostosis developing raised intracranial pressure from approximately 15% (in cases that do not have a genetic origin and do not have surgery) to less than 2% (in cases that have do have surgery). Children found to have a syndromic cause usually have a higher risk for developing raised intracranial pressure.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
This is the rarest of the major sutures to fuse and occurs in approximately 1 in 300,000 live births. The majority of cases affect only one lambdoid suture (unilambdoid) and so is more likely to have a genetic cause (such as ERF related craniosynostosis) and are most commonly caused by reduced space for the baby’s head in the uterus during pregnancy. Occasionally this affects both lambdoid sutures and is associated with fusion of other calvarial sutures and so is more likely to have a genetic cause. Unilambdoid synostosis leads to a flatness of the skull on the affected side of the back of the head together with a windswept appearance of the skull when viewed from both the front and the back. Surgery is in the form of a posterior remodelling procedure which usually takes place when affected children are around 12-18 months of age (though the widow of opportunity to undertake the surgery is quite wide). The aim of surgery is to stop the head shape from getting worse, make the head shape better by reshaping the back of the skull (occiput) and finally to reduce the risk of a child with lambdoid synostosis developing raised intracranial pressure from approximately 15% (in cases that do not have surgery) to less than 2% (in cases that have do have surgery) by creating more space around the brain. Children found to have a syndromic cause usually have a higher risk for developing raised intracranial pressure.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Syndromic Craniosynostosis
The vast majority of children with syndromic craniosynostosis have bicoronal synsotosis or multisuture synostosis. Some children with syndromic forms of craniosynostosis may require multiple operations throughout their childhood because of either the need for crisis intervention (acute urgent problems) or else for elective (non urgent) reasons. A crisis intervention may occur at any age in childhood and require surgery for any of the following reasons:
- development of raised intracranial pressure
- an urgent need to protect the eyes if there is severe exorbitism leading to exposure related damage to the cornea;
- severe sleep apnoea (breathing difficulties at night) due to significant underdevelopment of the midface which reduces airflow at the back of the nose.
Surgery for crisis intervention is complex and can take many forms depending on the cause of the problem and the circumstances of each individual syndromic case. Aside from the potential need for urgent craniofacial procedures to expand the skull and create more space for the brain there may also be a potential need for separate neurosurgical procedures e.g. to insert a shunt to drain an overproduction or blockage of fluid in the brain causing hydrocephalus or else to widen the opening at the base of the skull in the case of a Chiari-1 malformation (this is where the lowest portion of the back of the brain gets trapped and pinched in the bony opening at the base of the skull). Separate ENT procedures may also be required in extreme cases to protect the airway and improve breathing e.g. a tracheostomy.
The aim of primary elective surgery is to stop the head shape from getting worse, make the head shape better, and reduce the risk of a child with syndromic craniosynostosis developing raised intracranial pressure. This can take place by either surgery primarily aimed at reshaping the forehead (called a Frontoorbital Advancement and Remodelling procedure, or FOAR for short) or surgery to lengthen the back of the head in the first instance (called a Posterior Vault Distraction or PVD for short). Sometimes surgery is done to address the front and the back of the head at either the same time or else in a staged fashion.
Secondary elective surgery may involve midfacial advancement procedures as well as orbital dystopia correction (altering the position of the eye sockets if they are too far apart), forehead contouring, fat grafting, eyelid surgery, rhinoplasty (nose reshaping) and genioplasty (chin reshaping or repositioning). Sometimes these procedures are more appropriately performed when children are older and when their facial bone growth is mature but some of them can be performed at any age for crisis intervention.
Although all the syndromes are extremely rare, only the commoner syndromes are shown below. Some of the syndromes (particularly those with milder features) can look very similar and difficult to distinguish on clinical examination, and therefore require genetic testing to ultimately determine the correct diagnosis.
Click on the links below to find out more about each of these procedures:

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Treatment costs
Initial consultation
£300
The exact surgery cost will be confirmed after your consultation. This will include the hospital, surgeon and anaesthetic fees.
This procedure is available under a self-pay basis only.
Bicoronal synostosis refers to fusion of both coronal sutures and is much more likely to be genetic in origin than unicoronal synostosis or other single suture synostoses though some cases may still be caused by reduced space for the baby’s head in the uterus during pregnancy. The vast majority of syndromic craniosynostosis have bicoronal synsotosis. In bicoronal synostosis, the skull shape is very tall (called turricephaly) and broad and short from front to back with a flat forehead (called brachycephaly).
Surgery for bicoronal synostosis, can be either in one stage - in the form of a Frontoorbital Advancement and Remodelling procedure (FOAR) which usually takes place when affected children are around 12-18 months of age (though the widow of opportunity to undertake the surgery is quite wide), or else a 2 stage procedure with the first stage involving a Posterior Vault Distraction procedure (PVD) followed by an FOAR at least a year later. Either way, the aim of surgery is to stop the head shape from getting worse, make the head shape better and to reduce the risk of a child with bicoronal synostosis developing raised intracranial pressure. Of course depending on the syndromic cause, other surgical procedures may be required from childhood to maturity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Apert syndrome is a rare genetic syndromic form of craniosynostosis and occurs in 1 in 65,000 live births leading to a combination of craniofacial and limb abnormalities. The craniofacial features include brachycephaly (caused by bicoronal synostosis), shallow eye sockets (this is called exorbitism) making the eyes appear bulging and poor midfacial growth leading to an underdeveloped upper jaw and potential breathing problems such as sleep apnoea. Some cases have associated cleft palate. The limb features include complex symmetrical syndactyly (fusion of the fingers and toes). This requires surgery from a specialist hand surgeon affiliated to and working closely with the craniofacial team.
Apert syndrome is caused by one of 2 genetic mutations in a gene called the fibroblast growth factor receptor type 2 gene (or FGFR2 gene for short) giving rise to 2 different forms of Apert syndrome – one which has more complex facial features and less severe limb features, and one which has more severe limb features and less severe facial features. These genetic changes can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.
Individuals with Apert syndrome usually have learning difficulties and developmental delay, though there is a spectrum of severity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Crouzon syndrome is a rare genetic syndromic form of craniosynostosis and occurs in 1 in 60,000 live births. The craniofacial features (which can range from mild to severe) include brachycephaly (caused by bicoronal synostosis), shallow eye sockets (this is called exorbitism), making the eye appear bulging and poor midfacial growth leading to an underdeveloped upper jaw and potential breathing problems such as sleep apnoea. Some cases have associated cleft palate. The limbs are essentially normal in Crouzon syndrome.
The majority of cases of Crouzon syndrome are caused by genetic mutations in a gene called the fibroblast growth factor receptor type 2 gene (or FGFR2 gene for short). Rarely a specific mutation in the fibroblast growth factor receptor type 3 gene (or FGFR3 gene for short) can cause a more severe form of Crouzon syndrome. These genetic changes can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.
Individuals with Crouzon syndrome usually have normal intelligence.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Pfeiffer syndrome is a rare genetic syndromic form of craniosynostosis and occurs in 1 in 100,000 live births leading to a combination of craniofacial and limb abnormalities. The craniofacial features (which can range from mild to severe) include brachycephaly (caused by bicoronal synostosis), shallow eye sockets (this is called exorbitism), making the eye appear bulging and poor midfacial growth leading to an underdeveloped upper jaw and potential breathing problems such as sleep apnoea. Some cases have an associated cleft palate. In the severest form of Pfeiffer syndrome all the sutures (growth lines) of the skull and face can be prematurely fused leading to a cloverleaf appearance to the skull. The limb features typically include broad thumbs and big toes which are bent away from the rest of the hand and foot. Other limb features may also include short fingers and toes and webbing or fusion of the fingers and toes. Individuals with Pfeiffer syndrome often have learning difficulties and developmental delay, most commonly affecting speech and language.
The majority of cases of Pfeiffer syndrome are caused by genetic mutations in a gene called the fibroblast growth factor receptor type 2 gene (or FGFR2 gene for short). Rarely a specific mutation in the fibroblast growth factor receptor type 1 gene (or FGFR1 gene for short) gene can cause a milder form of Pfeiffer syndrome. These genetic changes can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.
Individuals with Pfeiffer syndrome often have learning difficulties and developmental delay, most commonly affecting speech and language, though there is a spectrum of severity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Saethre-Chotzen syndrome is a rare genetic syndromic and occurs in 1 in 50,000 live births leading to a combination of craniofacial and limb abnormalities. The craniofacial features include either plagiocephaly - an asymmetric forehead and skull being short from front to back on one side (caused by unicoronal synostosis) or brachycephaly – the skull being short from front to back on both sides (caused by bicoronal synostosis); shallow eye sockets (this is called exorbitism), making the eye appear bulging and poor midfacial growth leading to an underdeveloped upper jaw. There is also an association with drooping upper eyelids (called ptosis) and small low set ears.
Some cases have an associated cleft palate. The limb features may include some minor webbing of some of the fingers and toes which rarely require any surgery.
In most cases it is caused by a genetic mutation in a gene called the TWIST 1 gene in which case individuals have normal intelligence. In a smaller subset of cases, a complete deletion of the TWIST 1 gene along with neighbouring genes are the cause, and in these cases, the individual may have learning difficulties and developmental delay. This genetic change can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Muenke syndrome is a rare genetic syndromic and occurs in 1 in 30,000 live births. The craniofacial features include either plagiocephaly - an asymmetric flat forehead and skull being short from front to back on one side (caused by unicoronal synostosis) or brachycephaly – the skull being short from front to back on both sides with a flat and wide forehead (caused by bicoronal synostosis); shallow eye sockets (this is called exorbitism), making the eye appear bulging and poor midfacial growth leading to an underdeveloped upper jaw. This syndrome is associated with a type of hearing loss called sensorineural hearing loss. The craniofacial features in females are often more severe than in males. The limb features are usually mild and often do not require any surgical intervention.
Muenke syndrome is caused by a specific genetic mutation in a gene called the fibroblast growth factor receptor type 3 gene (or FGFR3 gene for short). This affects how the bone cells in the body grow. This genetic change can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
TCF12 related craniosynostosis syndrome is a newly identified genetic craniosynostosis syndrome. The craniofacial features most commonly cause brachycephaly – the skull being short from front to back on both sides (caused by bicoronal synostosis) but can also cause plagiocephaly - an asymmetric forehead and skull being short from front to back on one side (caused by unicoronal synostosis); shallow eye sockets (this is called exorbitism), making the eye appear bulging and poor midfacial growth. Clinical features overlap with those of Saethre Chotzen syndrome.
It is caused by a genetic mutation in a gene called the TCF12 gene. This genetic change can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.
A small number of children with TCF12 related craniosynostosis may have learning difficulties and developmental delay, though there is a spectrum of severity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
ERF related craniosynostosis syndrome is a newly identified genetic craniosynostosis syndrome accounting for around 2% of all syndromic cases of craniosynostosis. The craniofacial features include multiple suture synostosis with a combination of the sagittal and bilateral lambdoid sutures being the most predominantly affected and often occurring after birth with a delayed presentation and a delayed risk of developing raised intracranial pressure. Affected children may also have widely spaced eyes (hypertelorism), shallow eye sockets (exorbitism) and some underdevelopment of the midface. In addition affected children may exhibit a Chiari-1 malformation (this is where the lowest portion of the back of the brain gets trapped and pinched in the bony opening at the base of the skull).
It is caused by a genetic mutation in a gene called the ERF gene. This genetic change can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent, in which case there is a 50% chance of an affected parent passing on the affected gene to their children.
Some children with ERF related craniosynostosis have behavioural problems, speech and language delay, learning difficulties and developmental delay, though there is a spectrum of severity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk
Craniofrontonasal dysplasia is a rare genetic syndromic and occurs in 1 in 100,000 live births and affects girls more severely than boys. The craniofacial features include either plagiocephaly - an asymmetric flat forehead and skull being short from front to back on one side (caused by unicoronal synostosis) or brachycephaly – the skull being short from front to back on both sides with a flat and wide forehead (caused by bicoronal synostosis); shallow eye sockets (this is called exorbitism), making the eyes appear bulging. In addition the eye sockets in this condition are placed widely apart (called hypertelorism). There is often poor midfacial growth leading to an underdeveloped upper jaw. Girls tend to have frizzy hair, a groove in the middle of the nose while some cases have an associated cleft lip and palate. Additional features include sloping shoulders and some minor webbing of the fingers and toes which rarely require any surgery. Frequently longitudinal splits can be seen in the nails of the fingers and toes.
Craniofrontonasal dysplasia is caused by a genetic mutation in a gene called the EphrinB1 (EFNB1) gene on the X chromosome. This genetic change can either develop spontaneously (from unaffected parents) or else be inherited from an affected parent. Unlike most X-linked conditions girls are more severely affected while boys may not require any surgical intervention.
Individuals with Craniofrontonasal dysplasia may have some learning difficulties and developmental delay, though there is a spectrum of severity.

UK or International private referrals
Please contact private secretary and practice manager:
Marcia Pillai
Tel: 07780 514777
E-mail: davidjohnson@doctors.org.uk

































 Episode one: A Fighting Chance
Episode one: A Fighting Chance
 Episode two: Smile
Episode two: Smile
 Episode three: Rogue Gene
Episode three: Rogue Gene
